Theory
Quantum Chemistry: Probing the Electronic Structure of Molecules
Beyond Classical Force Fields
Classical molecular dynamics force fields are powerful tools for simulating large systems, but they treat atoms as point charges with fixed, pre-assigned properties. Bond lengths, angles, and partial charges are set once from experimental data or higher-level calculations and then frozen for the duration of the simulation. This approximation breaks down whenever we need to understand where electrons actually are — which determines chemical reactivity, charge distribution, and spectroscopic properties.
In principle, all molecular properties follow from solving the time-independent Schrödinger equation, $\hat{H}\psi = E\psi$, for the system’s wavefunction $\psi$. For a molecule with more than a handful of electrons, exact solutions are computationally intractable. Quantum chemistry provides a hierarchy of tractable approximations to this problem.
The dominant method in modern chemistry is Density Functional Theory (DFT). Rather than computing the full many-electron wavefunction, DFT works with the electron density $\rho(\mathbf{r})$ — a three-dimensional function describing how many electrons are present at each point in space. The Hohenberg-Kohn theorems (1964) proved that this density uniquely determines all ground-state properties of the system: the external potential, and hence the total energy. Kohn and Sham (1965) made this practical by introducing a fictitious system of non-interacting electrons that reproduces the same density as the real interacting system, transforming the many-body problem into a set of tractable single-particle equations.
In practice, DFT improves significantly on Hartree-Fock (HF) theory, which treats electron-electron repulsion through an average field and neglects correlation between electrons. DFT captures this correlation through an approximate exchange-correlation functional — a mathematical expression encoding all the complex quantum mechanical effects of electron-electron interactions. The choice of functional is the primary lever controlling accuracy.
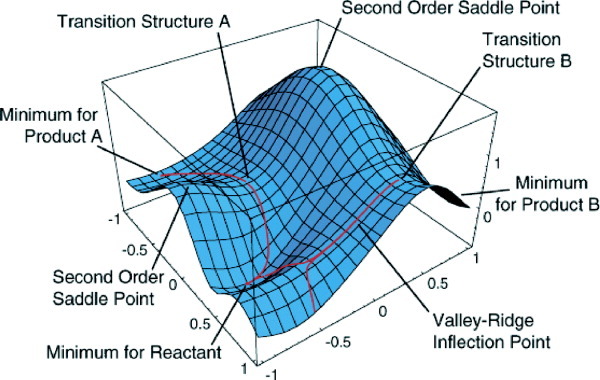
Figure 1: Potential Energy Surface (source: J. Comp. Chem.)
In drug discovery and the broader Ligand-X workflow, QM methods serve two key roles: geometry optimisation provides accurate 3D ligand structures for docking and molecular dynamics, and reactivity descriptors reveal electronic effects that classical force fields cannot capture. The central object for both is the Potential Energy Surface (PES), a landscape describing the total energy of the system as a function of all nuclear positions.
Geometry Optimization
A geometry optimization navigates the PES to find the most stable arrangement of atoms: a local minimum where the forces on every atom are zero. Mathematically, this is the point where the gradient of the energy ($\mathbf{g}$) vanishes.
The process is iterative: starting from an initial geometry (e.g., from a SMILES string), gradient-based algorithms like BFGS determine the direction and magnitude of each nuclear displacement. The calculation is considered converged when four quantities all fall below predefined thresholds: total energy change, maximum gradient, RMS gradient, and maximum step size.
Frequency Analysis
Geometry optimization finds a stationary point on the PES, but it doesn’t tell us whether that point is a stable minimum or a saddle point (transition state). A frequency calculation answers this by computing the Hessian matrix, the second derivatives of energy with respect to nuclear positions.
- All positive eigenvalues: A true energy minimum (a stable molecule)
- Exactly one negative (imaginary) frequency: A first-order saddle point—a transition state connecting two minima
Frequency calculations also yield the zero-point energy (ZPE), the vibrational energy a molecule retains at absolute zero, along with thermal corrections that convert raw electronic energy into Gibbs free energy (G), which is the quantity needed to assess thermodynamic spontaneity.
Predicting IR Spectra
Each vibrational mode identified by frequency analysis corresponds to a peak in the infrared (IR) spectrum, provided the vibration produces a change in the molecular dipole moment. The theoretical IR spectrum can be directly compared to experimental measurements to confirm a structure or identify unknowns.
In practice, standard harmonic approximations tend to overestimate vibrational frequencies by a few percent. Scaling factors (typically 0.95–0.98) are applied to correct for this systematic offset and improve agreement with experiment.
Fukui Indices: Mapping Reactivity
Conceptual DFT provides intuitive descriptors for predicting where a molecule is most likely to react. The Fukui function ($f$) identifies regions of electron density most susceptible to perturbation:
- $f^-$ (electrophilic attack): Sites most likely to be attacked by an electron-deficient reagent; regions dominated by HOMO density
- $f^+$ (nucleophilic attack): Sites most susceptible to attack by an electron donor; regions dominated by LUMO density
By condensing these 3D functions onto individual atoms, researchers obtain numerical rankings that predict regioselectivity in complex reactions: which site on a molecule gets functionalized first.
Choosing a Method
The accuracy of any QM calculation depends on two choices: the functional and the basis set.
Functionals range from simple Local Density Approximation (LDA) to sophisticated hybrid and double-hybrid functionals. The “gold standard” in quantum chemistry is CCSD(T) — coupled cluster theory with single, double, and perturbative triple excitations — which gives near-exact answers for small molecules at very high computational cost. For practical work on drug-sized molecules, hybrid DFT functionals such as B3LYP or PBE0 strike a good balance between cost and accuracy. Dispersion corrections (D3, D4) are essential for large organic molecules, where London dispersion forces significantly affect geometry and binding energies but are absent from standard functionals.
Basis sets define the mathematical space used to represent the electronic wavefunction as a linear combination of atom-centred functions. Reliable thermochemistry requires at least triple-ζ quality (e.g., def2-TZVP). Systems with anions or diffuse electron density need additional diffuse basis functions to describe the loosely bound electrons.
For rapid geometry optimisation of large organic molecules — for example, pre-processing a ligand library before docking — semiempirical methods such as GFN2-xTB offer DFT-quality geometries at a fraction of the cost by parameterising key integrals rather than computing them from first principles. This makes them practical for routine, high-throughput ligand preparation.
Quantum chemistry also underpins computational photochemistry: the nuclear ensemble approach used in the AtmoSpec workflow to calculate photoabsorption cross-sections relies on the same DFT framework to compute vertical excitation energies across a statistical ensemble of molecular geometries.
References
- Hohenberg, P., & Kohn, W. (1964). Inhomogeneous Electron Gas. Physical Review, 136(3B), B864–B871. doi:10.1103/PhysRev.136.B864
- Kohn, W., & Sham, L. J. (1965). Self-Consistent Equations Including Exchange and Correlation Effects. Physical Review, 140(4A), A1133–A1138. doi:10.1103/PhysRev.140.A1133
- Parr, R. G., & Yang, W. (1984). Density functional approach to the frontier-electron theory of chemical reactivity. Journal of the American Chemical Society, 106(14), 4049–4050. doi:10.1021/ja00326a036
- Grimme, S., Ehrlich, S., & Goerigk, L. (2011). Effect of the damping function in dispersion corrected density functional theory. Journal of Computational Chemistry, 32(7), 1456–1465. doi:10.1002/jcc.21759
- Weigend, F., & Ahlrichs, R. (2005). Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Physical Chemistry Chemical Physics, 7(18), 3297–3305. doi:10.1039/B508541A
- Becke, A. D. (1993). Density-functional thermochemistry. III. The role of exact exchange. Journal of Chemical Physics, 98(7), 5648–5652. doi:10.1063/1.464913
- Neese, F. (2012). The ORCA program system. WIREs Computational Molecular Science, 2(1), 73–78. doi:10.1002/wcms.81
- Grimme, S., Bannwarth, C., & Shushkov, P. (2017). A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All spd-Block Elements. Journal of Chemical Theory and Computation, 13(5), 1989–2009. doi:10.1021/acs.jctc.7b00118
Konstantin N
BLOG
Quantum Chemistry PES DFT Theory