
Theory
Input Sanitization: Preparing for Simulation
Why Preparation Matters
The reliability of any molecular simulation is only as good as the input structure. Experimental protein structures are deposited in the Protein Data Bank (PDB), a freely accessible repository of over 200,000 experimentally determined 3D structures. Each entry is stored as a .pdb file containing the Cartesian coordinates of every non-hydrogen atom resolved by the experiment. Similarly, small molecules are often encoded as SMILES strings (Simplified Molecular Input Line Entry System) — a compact text notation that encodes atomic connectivity, such as CCO for ethanol — which must be converted to a 3D structure before simulation.
Neither of these formats is simulation-ready out of the box. X-ray crystallography, for instance, often fails to resolve hydrogen atom positions, flexible loop regions, or the correct protonation states of titratable residues. Running a simulation on an uncleaned structure is a guaranteed path to bad results, which is why preparation is treated as a disciplined, systematic process.
Protein Refinement
Raw PDB structures frequently have defects that need to be addressed before any physics-based calculation can proceed:
Structural completeness: Utilities like PDBFixer detect and repair missing atoms and residues. A discontinuous protein backbone, caused by unresolved loops, would produce physically unrealistic behavior during dynamics, so loop reconstruction is essential.
Protonation states: Assigning correct hydrogen atoms is perhaps the most chemically significant step. The protonation of titratable residues like Histidine, Aspartate, and Glutamate depends on the local electrostatic environment, which can shift their effective pKa away from standard solution values. This matters because protonation state determines charge: a protonated aspartate is neutral, while a deprotonated one carries a −1 charge. A misassigned charge changes the electrostatic interactions within the binding pocket and can distort the protein’s three-dimensional structure during simulation. Histidine alone can exist in two neutral tautomeric forms (HID or HIE, differing in which nitrogen bears the proton) or a positively charged form (HIP); an incorrect assignment is one of the most common causes of simulation artefacts.
Chemical cleaning: Experimental structures often include crystallization artifacts: non-biological ligands, buffer molecules, and ions (glycerol, PEG, etc.) that must be removed before simulation to avoid interfering with the biology of interest.
Ligand Preparation
Small molecule ligands often begin as a 1D SMILES string: a compact encoding of connectivity that lacks 3D coordinates.
Figure 1: Benzene molecule
3D coordinate generation: The SMILES must first be converted to a full molecular graph (atoms as nodes, bonds as edges) with all implicit hydrogens made explicit. Conformational sampling then identifies low-energy 3D geometries, including different ring puckers and rotameric states.
Ionization and tautomers: Biological activity depends on the dominant ionization state at physiological pH (7.4). Software like Dimorphite-DL calculates pKa values and assigns appropriate charge states. Selecting the wrong tautomer or ionization state can silently corrupt downstream docking or free energy results.
System Integration and Relaxation
Once both the protein and ligand are individually prepared, they must be assembled into a single simulation system. This requires combining their respective force field topologies — files that define the atom types, partial charges, and bonding connectivity that the simulation engine will use. Different force fields are used for different components: ff14SB is a widely used parameter set for proteins, while a specialized force field such as GAFF2 (General Amber Force Field 2) or OpenFF Sage, designed for small organic molecules, both having been parameterised to reproduce experimental geometries and thermodynamic data.
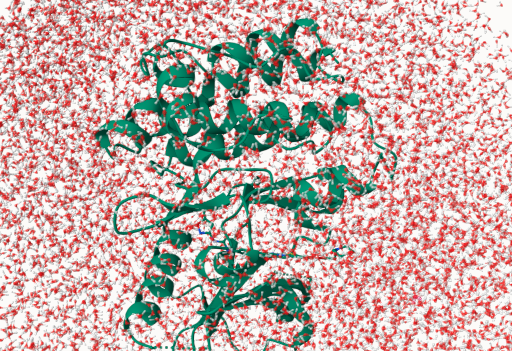
Figure 2: Solvated FLT3 system with a small molecule inhibitor
The combined system is then placed in a periodic box of explicit water molecules. The most commonly used water model is TIP3P (Transferable Intermolecular Potential with 3 Points), which represents each water molecule as three point charges — one oxygen and two hydrogens — at experimental geometry. The box must include a sufficient buffer of water around the solute (typically ~10 Å) on all sides. Periodic boundary conditions (PBC) are applied so that atoms leaving one face of the box re-enter from the opposite face, effectively mimicking an infinite bulk environment and eliminating artificial surface effects. Counterions (Na⁺, Cl⁻) are added to neutralise the total charge and reach physiological salt concentration (~150 mM).
Finally, a tiered relaxation protocol prepares the system for production:
- Energy minimization: Resolves steric clashes — atoms that accidentally overlap during system assembly — using gradient-based algorithms such as Steepest Descent
- NVT equilibration: Heats the system to the target temperature at constant Number of particles, Volume, and Temperature
- NPT equilibration: Adjusts pressure and density at constant Number of particles, Pressure, and Temperature, stabilising the box dimensions before data collection begins
The quality of this preparation determines the reliability of every downstream measurement. As the principle goes: garbage in, garbage out.
With a clean, fully protonated, and solvated system in hand, we are now ready to ask our first scientific question: where does this ligand bind, and how strongly? That is the domain of molecular docking.
References
- Eastman, P., et al. (2017). OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLOS Computational Biology, 13(7), e1005659. doi:10.1371/journal.pcbi.1005659
- Ropp, P. J., Kaminsky, J. C., Yablonski, S., & Durrant, J. D. (2019). Dimorphite-DL: An open-source program for enumerating the ionization states of drug-like small molecules. Journal of Cheminformatics, 11(1), 14. doi:10.1186/s13321-019-0336-9
- Maier, J. A., et al. (2015). ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. Journal of Chemical Theory and Computation, 11(8), 3696–3713. doi:10.1021/acs.jctc.5b00255
- Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., & Case, D. A. (2004). Development and testing of a general amber force field. Journal of Computational Chemistry, 25(9), 1157–1174. doi:10.1002/jcc.20035
- Landrum, G. et al. RDKit: Open-Source Cheminformatics. rdkit.org — the standard open-source toolkit for 3D coordinate generation and ligand preparation.
- Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., & Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics, 79(2), 926–935. doi:10.1063/1.445869
Konstantin N
BLOG
Protein Simulation Theory